pKALM: Accurate and Rapid Protein pKa Prediction from Sequence
pKALM[1] predicts protein pKa values directly from amino-acid sequence using a protein language model and transfer learning — no structure required. It reaches state-of-the-art accuracy (0.8658 RMSE) at a throughput of ~4,965 pKa/s, covering six ionizable side chains (Asp, Glu, His, Lys, Cys, Tyr) plus the N- and C-termini.
Run a Prediction
Paste one or more sequences in FASTA format or upload a FASTA file, pick a model, and submit. Each run opens a dedicated, bookmarkable result page.
Predictions currently run on a CPU server. pKALM is sequence-based and fast, so most jobs finish in seconds to a minute. The result page shows live progress, can be bookmarked or shared, and is retained for one week.
How pKALM Works
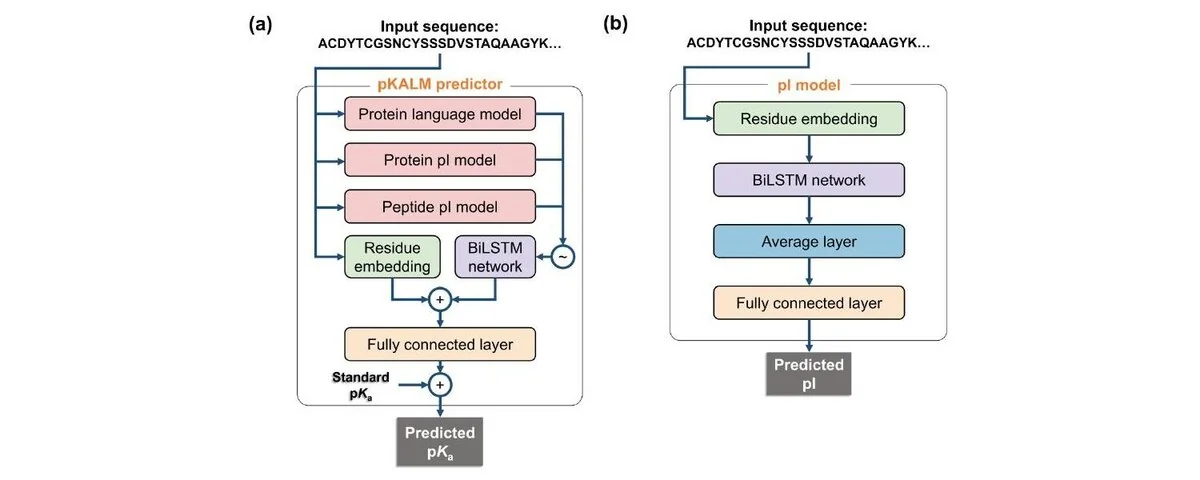
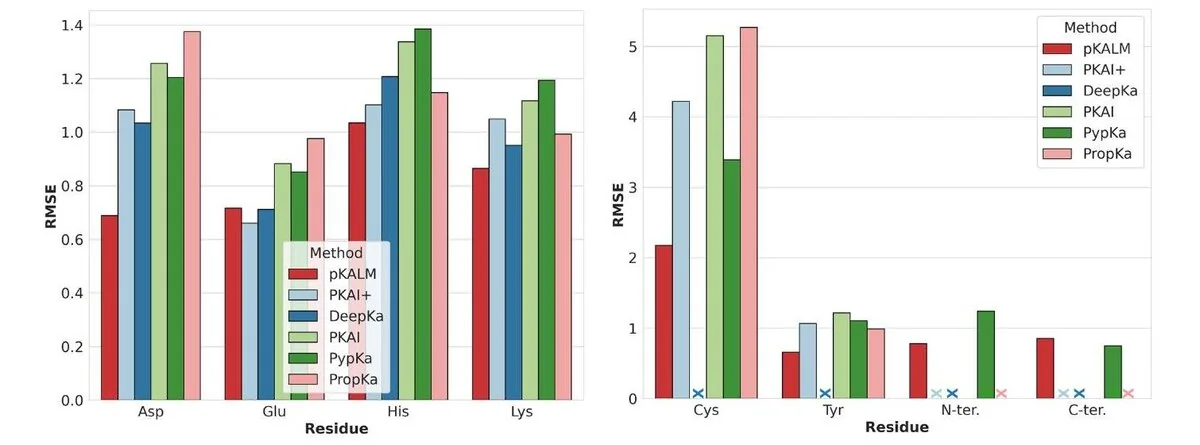
Performance
pKALM is benchmarked against widely used pKa predictors — the empirical method PROPKA[3], the Poisson–Boltzmann solver PypKa[4], and the deep-learning methods pKAI / pKAI+[5] and DeepKa[6].
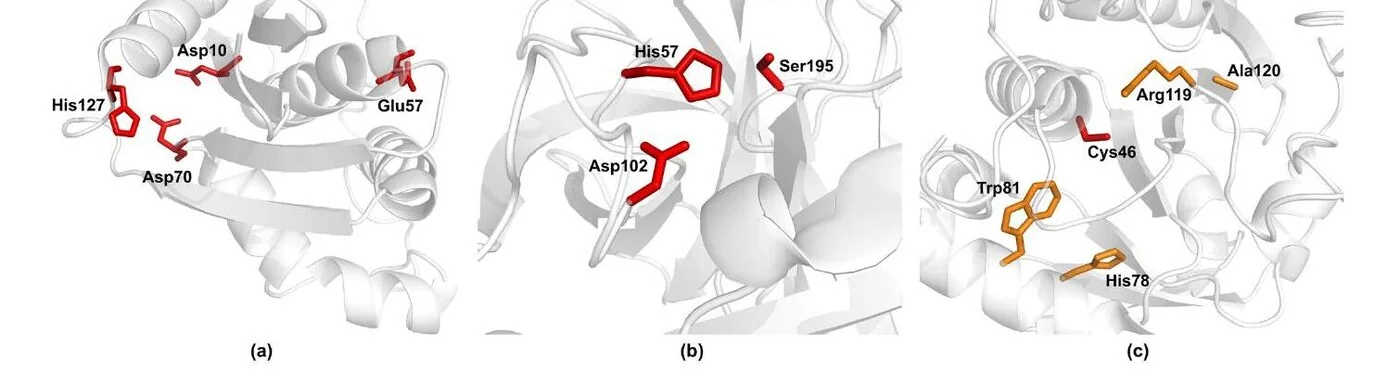
Case Studies
Beyond aggregate metrics, pKALM recovers chemically meaningful sites directly from sequence — including catalytic residues whose pKa is strongly perturbed by their environment.
Supported Ionizable Groups
Each job also returns predicted isoelectric points (pI) for the peptide and protein models, appended to the results.
References
- ^ Xu, S.; Onoda, A. Accurate and Rapid Prediction of Protein pKa: Protein Language Models Reveal the Sequence–pKa Relationship. J. Chem. Theory Comput. 2025, 21 (7), 3752–3764. DOI: 10.1021/acs.jctc.4c01288
- ^ Lin, Z.; Akin, H.; Rao, R.; Hie, B.; Zhu, Z.; Lu, W.; Smetanin, N.; Verkuil, R.; Kabeli, O.; Shmueli, Y.; et al. Evolutionary-Scale Prediction of Atomic-Level Protein Structure with a Language Model. Science 2023, 379 (6637), 1123–1130. DOI: 10.1126/science.ade2574
- ^ Olsson, M. H. M.; Søndergaard, C. R.; Rostkowski, M.; Jensen, J. H. PROPKA3: Consistent Treatment of Internal and Surface Residues in Empirical pKa Predictions. J. Chem. Theory Comput. 2011, 7 (2), 525–537. DOI: 10.1021/ct100578z
- ^ Reis, P. B. P. S.; Vila-Viçosa, D.; Rocchia, W.; Machuqueiro, M. PypKa: A Flexible Python Module for Poisson–Boltzmann-Based pKa Calculations. J. Chem. Inf. Model. 2020, 60 (10), 4442–4448. DOI: 10.1021/acs.jcim.0c00718
- ^ Reis, P. B. P. S.; Bertolini, M.; Montanari, F.; Rocchia, W.; Machuqueiro, M.; Clevert, D.-A. A Fast and Interpretable Deep Learning Approach for Accurate Electrostatics-Driven pKa Predictions in Proteins. J. Chem. Theory Comput. 2022, 18 (8), 5068–5078. DOI: 10.1021/acs.jctc.2c00308
- ^ Cai, Z.; Luo, F.; Wang, Y.; Li, E.; Huang, Y. Protein pKa Prediction with Machine Learning. ACS Omega 2021, 6 (50), 34823–34831. DOI: 10.1021/acsomega.1c05440
Please contact shijie.xu@ees.hokudai.ac.jp for any questions.
Changelogs
- 2026-05-28Rebuilt the web interface with a live result page.
- 2025-11-09Updated citation.
- 2024-10-22First release.