PsiPartition: Improved Site Partitioning for Genomic Data by Parameterized Sorting Indices and Bayesian Optimization
PsiPartition[1] is a tree-independent site-partitioning method for phylogenetic analysis. It scores every site with a Parameterized Sorting Index (PSI) and uses Bayesian optimization to automatically determine the best partitioning scheme — without requiring a reference tree or prior knowledge of the data. This yields more accurate reconstructions, especially for large genomic datasets with strong site heterogeneity. [ GitHub | Paper ]
Introduction
DNA (deoxyribonucleic acid) is the blueprint of life. It carries the instructions to make proteins, which are essential for all living things. These instructions are written using a code made of three-letter "words" called codons (Figure 1). The same codon code is used by almost all life forms, showing how all living things are connected. However, the code is not one-to-one, meaning that multiple codons can code for the same amino acid. This redundancy is called "degeneracy" and is thought to be a result of evolution. The difference of code for the same amino acids usually happens at the third position of the codon, which is called the "synonymous" site. In contrast, the first and second positions are called "non-synonymous" sites. Such difference means that the third position is less important for the protein structure and function, and is under less selective pressure.

The difference in selective pressure between synonymous and non-synonymous sites are considered in the partitioned models in phylogenetic inference. These models assume that different sites in the sequence alignment have different evolutionary rates, and use different substitution matrices for different sites. The partitioned models can improve the accuracy of phylogenetic inference, especially for large genomic data with more site heterogeneity. However, the partitioned models require the user to specify the number of partitions and the sites in each partition. This is a challenging task, as the user needs to have prior knowledge of the data to make the partitioning.
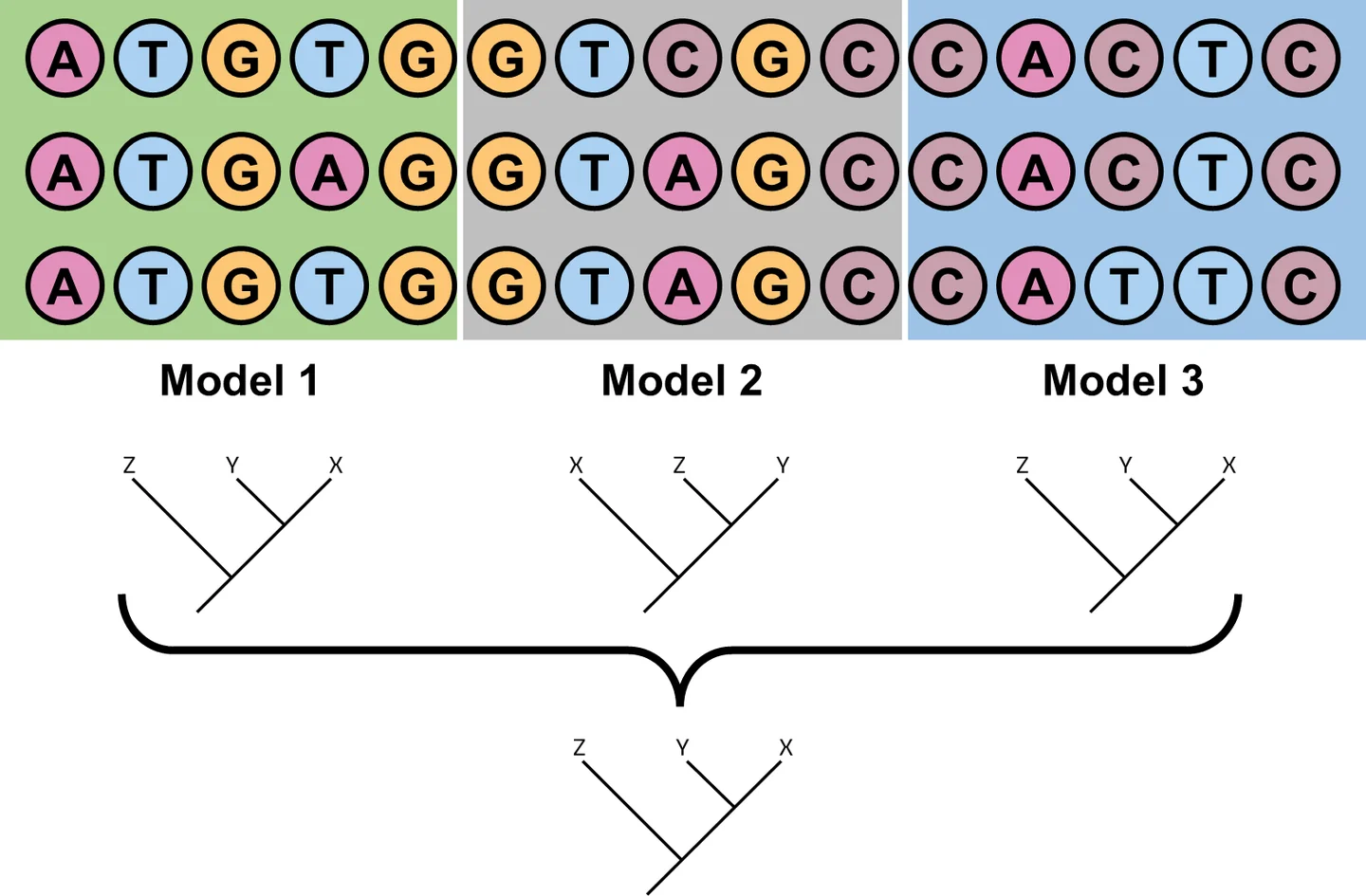
How PsiPartition Works
The hard part of a partitioned model is deciding how many partitions to use and which sites go into each one. The number of possible partitionings of an alignment grows faster than exponentially with the number of sites (the Bell number), so an exhaustive search is impossible. Existing automatic methods either depend on a reconstructed reference tree — which is slow to obtain and may itself be wrong — or rely on greedy search that can get trapped in local optima.
PsiPartition avoids both problems with two ideas:
- Parameterized Sorting Index (PSI). Building on the tree-independent TIGER rate estimator[2], PsiPartition assigns each site a score that measures how much its pattern of characters agrees with the rest of the alignment — without ever building a tree. Two refinements make it more expressive: a set of learnable weights that let different characters (bases, amino acids, gaps) contribute differently, and a pairwise comparison with a small noise term that prevents all conserved (invariant) sites from being dumped into a single partition, which would bias the inferred tree.
- Bayesian optimization. The number of partitions k and the weights are chosen automatically by Bayesian optimization, which fits a Gaussian-process surrogate of the objective (the model's BIC, evaluated with IQ-TREE[3]) and proposes promising settings to try next. Because there are only a few parameters and each evaluation is expensive, this is far more efficient than greedy search. A lighter variant, PsiPartitionFast, fixes the weights and optimizes only k.
Sites are then sorted by their PSI and binned into the chosen number of partitions, so slowly-evolving conserved sites and fast-evolving variable sites end up in different partitions.
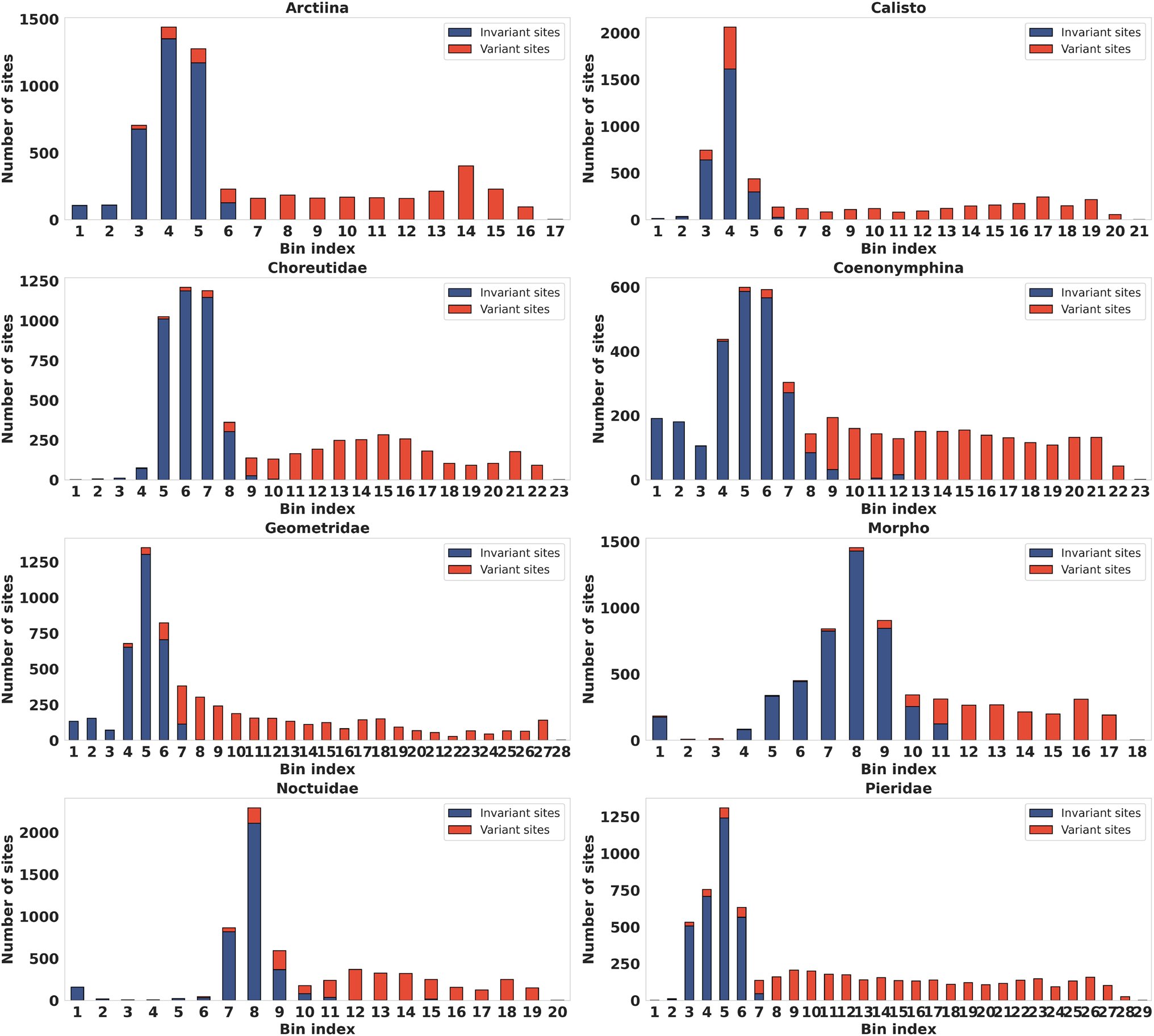
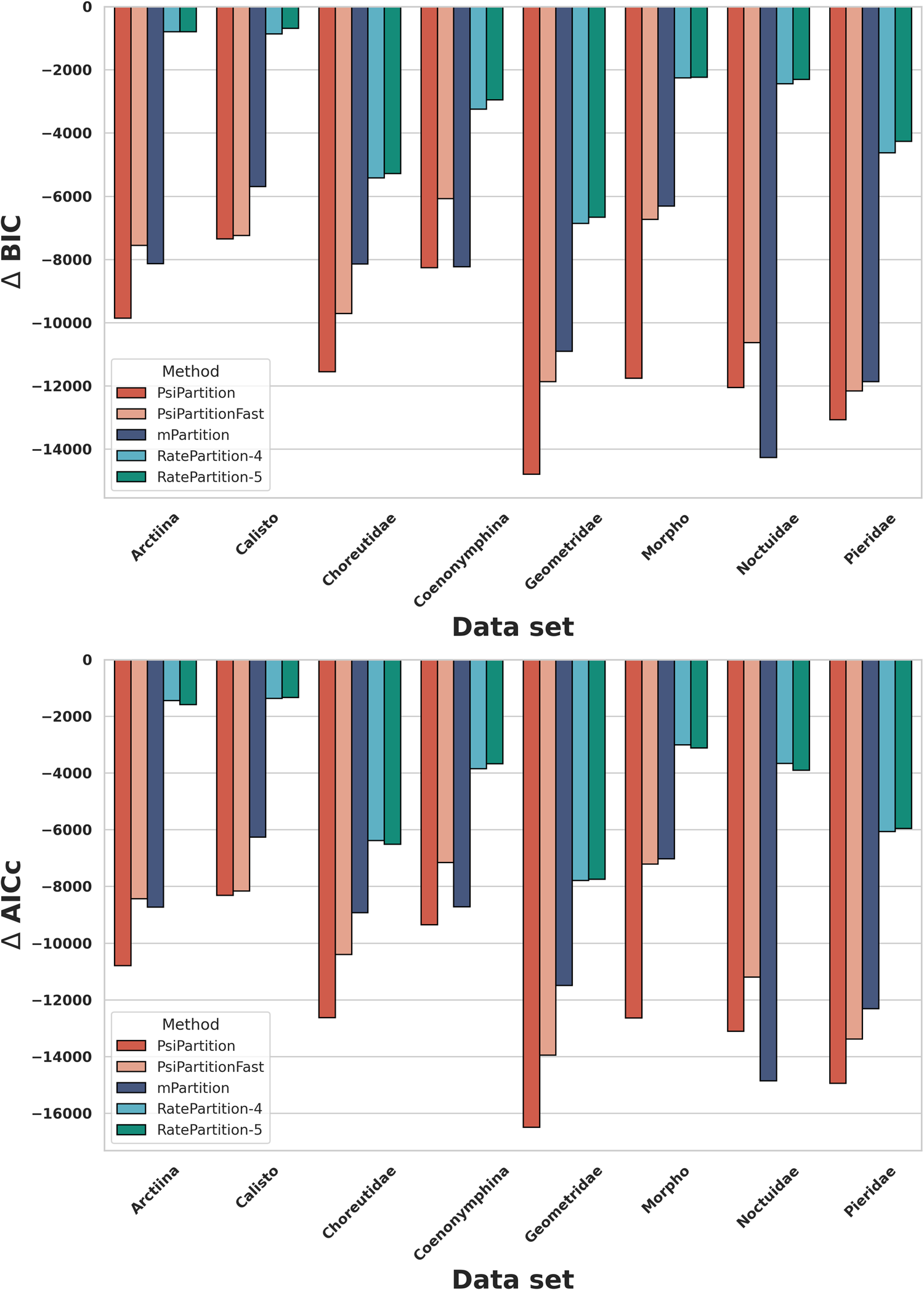
Performance
Across eight empirical DNA datasets, PsiPartition and PsiPartitionFast fit the data substantially better than existing partitioning methods (mPartition[4], RatePartition[5]), giving much lower Bayesian Information Criterion (BIC) and corrected Akaike Information Criterion (AICc) values.
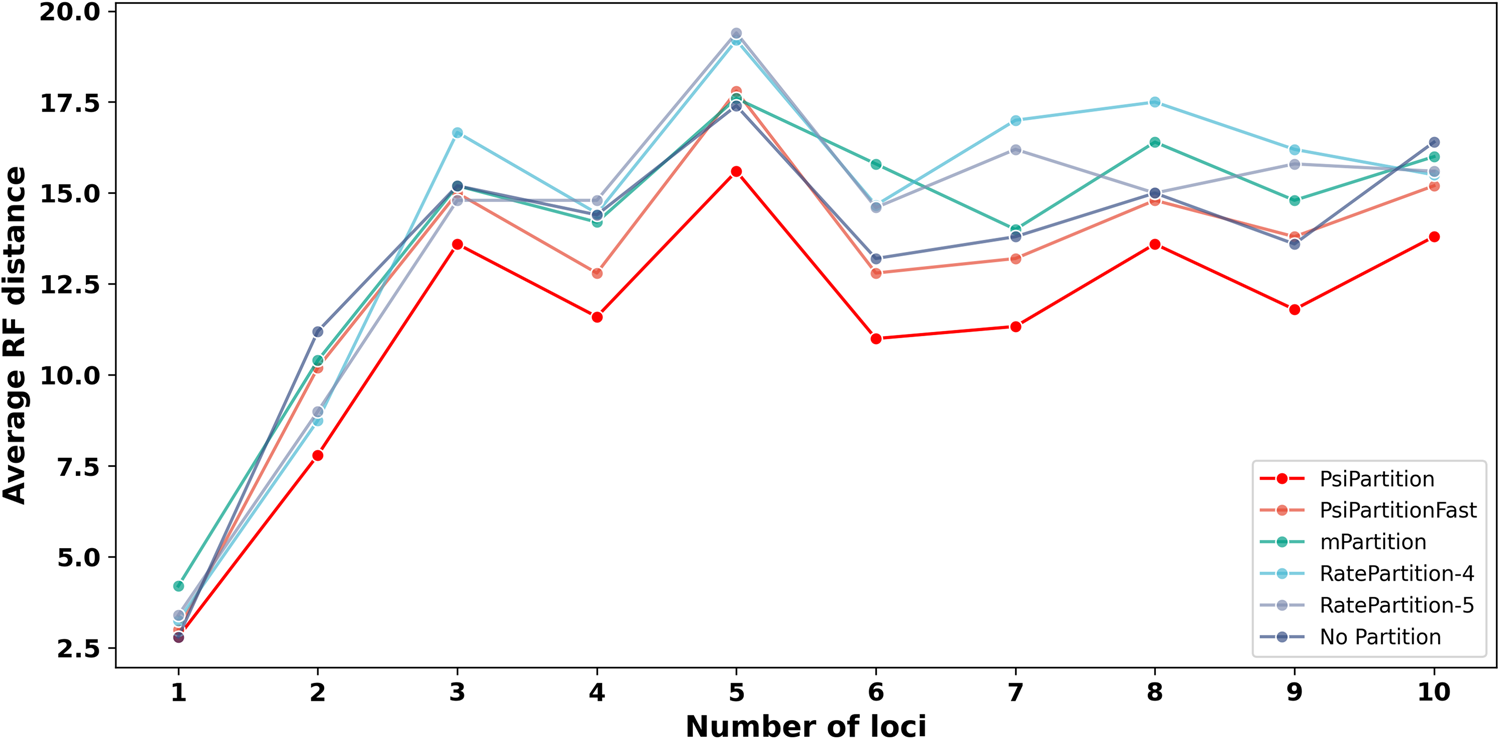
More importantly, better model fit translates into more accurate trees. On simulated data with heterogeneous evolutionary rates, PsiPartition reconstructs the trees closest to the truth — the smallest Robinson–Foulds (RF) distance[6] — and its advantage grows as the number of loci increases. It also outperforms existing methods on empirical protein datasets.
How to Use PsiPartition
Step 1: Preparation
Before using PsiPartition, please prepare the following things:
- A sequence alignment in FASTA format. Suppose you have a query sequence and you want to infer its phylogenetic relationship with other sequences. You need to search for homologous sequences from the database such as UniProt and align them by using software such as COBALT.
- Phylogenetic software. PsiPartition is based on partitioned models, and it does not perform phylogenetic inference itself. We use IQ-TREE as our host software. Click the link and unzip the files into some folder. You can test if it works by running:
./bin/iqtree2.exe -s example.phy
Figure 6: IQ-TREE phylogenetic software. The software is used to infer phylogenetic trees from sequence alignments. - Python. PsiPartition is written in Python, so you need to have Python installed on your computer. You can download Python from here.
- PsiPartition. Download the PsiPartition software from here. Unzip it somewhere, go to the folder and install required packages:
pip install -r requirements.txt
- A Weights & Biases account. PsiPartition uses Weights & Biases to log the optimization process. You need to sign up for an account and get your API key.
Step 2: Run PsiPartition
After you have prepared the above things, you can run PsiPartition by following the command below:
python PsiPartition_wandb.py --msa MSA_File --format fasta --alphabet dna --max_partitions 5 --n_iter 100
The arguments are:
--msa: The path to the sequence alignment file in FASTA format.--format: The format of the alignment file. It can be eitherfastaorphylip.--alphabet: The alphabet of the sequences. It can be eitherdnaoraa.--max_partitions: The maximum number of partitions to be optimized.--n_iter: The number of iterations for Bayesian optimization.
The *.iqtree file contains the reconstructed phylogenetic tree. You can visualize the tree using software such as iTOL. In addition, the file *.parts contains the optimized partitioning scheme. You can use this file to analyze the data with partitioned models in IQ-TREE:
./bin/iqtree2.exe -s example.phy -spp example.parts
References
Users are kindly requested to utilize the following citation when referencing this method:
- ^ Xu, S.; Onoda, A. PsiPartition: Improved Site Partitioning for Genomic Data by Parameterized Sorting Indices and Bayesian Optimization. J. Mol. Evol. 2024. DOI: 10.1007/s00239-024-10215-7
- ^ Cummins, C. A.; McInerney, J. O. A Method for Inferring the Rate of Evolution of Homologous Characters That Can Potentially Improve Phylogenetic Inference, Resolve Deep Divergence and Correct Systematic Biases. Syst. Biol. 2011, 60 (6), 833–844. DOI: 10.1093/sysbio/syr064
- ^ Minh, B. Q.; Schmidt, H. A.; Chernomor, O.; Schrempf, D.; Woodhams, M. D.; von Haeseler, A.; Lanfear, R. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 37 (5), 1530–1534. DOI: 10.1093/molbev/msaa015
- ^ Le, T. K.; Le, V. S. mPartition: A Model-Based Method for Partitioning Alignments. J. Mol. Evol. 2020, 88 (8–9), 641–652. DOI: 10.1007/s00239-020-09963-z
- ^ Rota, J.; Malm, T.; Chazot, N.; Peña, C.; Wahlberg, N. A Simple Method for Data Partitioning Based on Relative Evolutionary Rates. PeerJ 2018, 6, e5498. DOI: 10.7717/peerj.5498
- ^ Robinson, D. F.; Foulds, L. R. Comparison of Phylogenetic Trees. Math. Biosci. 1981, 53 (1–2), 131–147. DOI: 10.1016/0025-5564(81)90043-2
Please contact shijie.xu@ees.hokudai.ac.jp for any questions.
Changelogs
- 2026-05-27Expanded the method and performance sections with figures from the paper.
- 2024-12-16Added the guide on how to use PsiPartition.
- 2024-08-08First release.